Cystic fibrosis
Also known as mucoviscidosis
Key points about cystic fibrosis (CF)
- Cystic fibrosis (CF) is an inherited, lifelong genetic condition.
- CF is the most common life threatening genetic condition in Aotearoa New Zealand and affects more than 600 New Zealanders.
- CF affects the lungs, liver, digestive system and other parts of the body.
- Management of CF includes physiotherapy, exercise and medicines.
- Recent advances in medicines mean that many people with CF now have a much brighter future.

Cystic fibrosis is caused by a faulty gene that has been passed down from both a baby's mum and dad and is usually diagnosed soon after birth. Occasionally, however, CF is diagnosed in adolescence or adulthood.
Genes are what make us who we are – they affect our eye colour, hair colour, how tall we are and many of the things that make us individuals. Genes also affect how our body works on the inside, and changes in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene causes CF.
Every person has 2 copies of their genes – 1 from their father and 1 from their mother. Many people carry 1 copy of the CFTR gene (often called the CF gene) but you need 2 copies to be born with CF. Having 1 copy (being a carrier) doesn’t affect your health. If 2 people who carry a CF gene have a child together, there is a 1 in 4 chance that the baby will be born with CF.
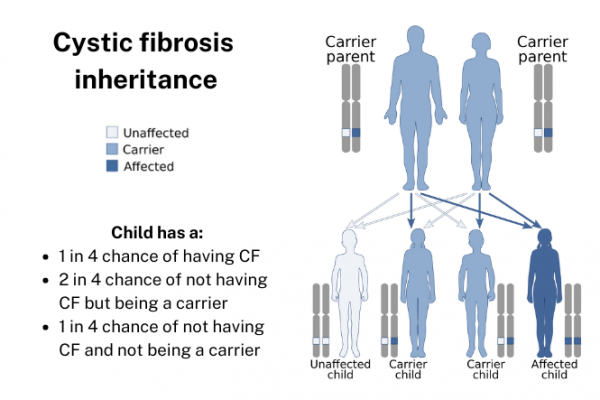
Image credit: Domaina, Kashmiri and SUM1 via Wikimedia Commons(external link) and Healthify He Puna Waiora
There are many gene mutations that cause CF, the most common being the F508del mutation. Approximately 90% of people with CF in Aotearoa New Zealand carry at least 1 copy of the F508del mutation.
Most babies born in Aotearoa New Zealand are screened for CF at birth and are generally diagnosed before any symptoms develop. If a child has missed routine screening, symptoms at birth can include:
- salty tasting skin
- slow weight gain or failure to thrive even with a good appetite
- wheezing, coughing and pneumonia
- abnormal bowel movements.
For a small number of babies, the first symptom of CF is a blocked intestine at birth, known as meconium ileus. Meconium is the thick, sticky black secretion from a baby’s bowel, usually passed within 24 hours of birth. In some babies with CF, the meconium is too thick to pass and blocks their intestine. They need enemas (a laxative solution put into their bottom) or surgery to remove it.
When we don’t have CF, our bodies make mucus that is thin and slippery and works as a lubricant to help protect us from infections and to keep the inside of our body working well. If we have CF, our mucus becomes thick and sticky and blocks the tiny tubes of many of our organs.
CF causes the body to produce thick, sticky mucus which affects the lungs, liver, digestive system and other parts of the body. The type and severity of symptoms varies between individuals – some people with CF remain well for a long period of time with minimal symptoms or hospital admissions, while others require more intensive medical care.
Lungs
In your lungs, the sticky secretions are difficult to cough up and viruses, bacteria and fungus can become trapped under the mucus. This can cause inflammation and infection, often causing a chronic cough and repeated chest infections. People with CF also have a much higher risk of developing serious complications from bugs that are generally harmless to other people. Most people with CF will develop some lung damage over time.
Digestive system
In your digestive system, thick secretions also block the flow of digestive enzymes from the pancreas to the duodenum (top part of the intestine), where they’re needed to help break down food. Without these enzymes, fats and vitamins can’t be absorbed. This causes problems with malnutrition and poor weight gain.
The lack of digestive enzymes in the duodenum caused by thick mucus causes:
- malnutrition due to malabsorption
- poor weight gain
- frequent, foul-smelling, greasy bowel motions
- stomach aches
- excessive wind.
Some people with CF may also develop cystic fibrosis related diabetes (CFRD).
Read more about nutrition and cystic fibrosis(external link).
Most babies in Aotearoa New Zealand are tested for CF through the Newborn Metabolic Screening Programme, often referred to as the Guthrie Heel Prick test. This test, usually carried out about 48 hours after birth, uses a blood sample taken from a baby’s heel and screens it for rare disorders, including CF.
If the results show the baby may have CF, a second heel prick test will be done. Other tests of the baby’s sweat, poo and blood will be done to confirm the diagnosis. Read more about how CF is diagnosed(external link).
Every day, usually twice a day, a person with CF needs to do chest physio to help keep their lungs clear of mucus. This can take 20 to 40 minutes each time but it can be longer if they're unwell. Chest physio is usually done in the morning, for example before school or work, and before bed.
Read more about physiotherapy for CF(external link).
If you have advanced cystic fibrosis, you may need to have a lung transplant. A lot of testing and assessment is needed to make sure you’re eligible. Read more about lung transplantation(external link).
Most people with CF need to take pancreatic enzyme capsules with meals and snacks to help them digest their food and absorb nutrients, eg, Creon®.
Medicines used in the treatment of CF include:
- antibiotics
- bronchodilators
- vitamin supplements
- insulin for those with cystic fibrosis-related diabetes CFRD.
CFTR Modulator Therapies
Until recently, medicines for CF treated only the symptoms of this condition. However, there are now medicines called CFTR modulator therapies that treat the cause of CF by correcting the defects in the CF gene. Two CFTR modulator therapies are now publicly funded in Aotearoa New Zealand.
One of these modulator therapies (Trikafta) targets the defects in the F508del CF gene mutation but also works for a wide range of other mutations. It's currently funded in Aotearoa New Zealand for people with CF who have eligible mutations and who are 6 years of age and older.
Trikafta has been shown to provide significant improvements in lung function, weight gain, quality of life and increased life expectancy, and may help to delay the progression of the condition in younger people. CTFR modulator therapies have changed the lives of many people with CF providing them with a much brighter future.
Read about how CF is managed with treatment and medication(external link).
Read more about CFTR modulator therapies(external link).
Cystic Fibrosis Association of New Zealand(external link)(external link)
Freephone 0800 651 122. Support for people with CF and their families – through a free library, grants and awards, social networks, newsletters, educational opportunities, fundraising and more.
Cystic Fibrosis NZ(external link)
Cystic fibrosis frequently asked questions(external link) Cystic Fibrosis NZ
Brochures
Fact sheets(external link) Cystic Fibrosis NZ
Your newborn baby's blood test – the newborn metabolic screening programme(external link) HealthEd, NZ, 2025
References
Standards of care for cystic fibrosis in New Zealand(external link) Medical Advisory of Cystic Fibrosis Association of New Zealand, 2010
Newborn screening and diagnostic protocol for cystic fibrosis in NZ(external link) National Screening Unit, NZ, 2016
Australasian clinical practice for social work in cystic fibrosis 2017(external link) Australian Cystic Fibrosis Social Work Interest Group
More detail about investigations, diagnosis and treatment options(external link) Patient Info, UK
Cystic Fibrosis NZ(external link)
Child and youth cystic fibrosis clinical network(external link) Starship, NZ
Brochures

Cystic Fibrosis New Zealand

{kind=link}
Need help now?

![]()
![]()
Credits: Healthify editorial team, based on content from Cystic Fibrosis NZ. Healthify is brought to you by Health Navigator Charitable Trust.
Reviewed by: Cystic Fibrosis NZ
Last reviewed:
